基因组序列是决定生物体生物学特性和演化历史的关键,而从头组装(De novo assembly)是从测序到重建生物体基因组序列的过程,这一问题在生物信息学中已存在四十年。传统上,基因组通常被组装成几兆基(Mb)的碎片段,但随着长读长测序技术的进步,现在对许多生物体而言,能够实现接近完整染色体的组装,即端粒到端粒(telomere-to-telomere)的组装。
4月22日,顶级生物信息学家李恒教授在Nature Review Genetics(IF=42.69)发表了题为“Genome assembly in the telomere-to-telomere era”的综述论文,该综述详细总结了目前T2T组装的发展过程,重点讨论了如何获得接近端粒到端粒的组装,并展望了解决剩余组装间隙和组装非二倍体基因组所需的技术和算法进步。

图1 文章发表信息
Part 1
T2T组装所需的测序数据类型
当前,二倍体T2T组装主要依靠长读长数据结合其他多种数据类型以解决重复序列和不同长度尺度的分型问题。长读长测序技术,如PacBio的HiFi reads和牛津纳米孔技术(ONT),能够产生通常≥10 kb长度的连续读长序列。PacBio在2019年推出的HiFi reads长度为10-20 kb,错误率低于0.5%,成为高质量组装的核心数据类型。为了接近T2T组装,还需要结合≥100 kb长度的ONT超长读长数据,尽管它们的准确性较低(大约95%),但有助于解决HiFi reads单独无法组装的剩余重复序列。
Hi-C是一种广泛使用的长距离数据类型,它通过揭示空间上接近的位点间的相互作用来帮助将contigs辅助组装到染色体上。其他可用于辅助组装的数据类型包括Pore-C(ONT+Hi-C)、Strand-seq,以及现代短读长技术,如stLFR、TELL-seq和haplotagging(它们产生来自基因组约100 kb片段的短读长簇,成本较低)。BioNano光学限制图谱也提供了长距离信息,但它不生成连续的核苷酸读长,无法解决超过读长长度的重复序列,因此在T2T组装中的适配性不如HiFi+ONT+Hi-C。
表1 T2T组装所需的测序数据类型

Part 2
T2T基因组的组装算法
当前实现T2T基因组组装的流程大致分为四个关键步骤:首先,对准确的长读长序列进行错误校正;其次,基于校正后的序列构建组装图(assembly graph);接着,利用超长序列对组装图进行简化;最后,使用Hi-C进行分型和辅助组装(phasing and scaffolding)。
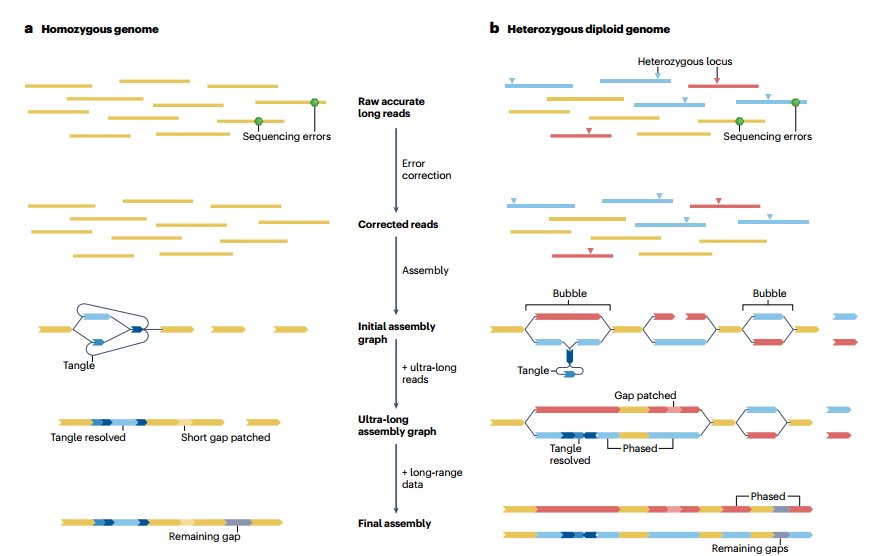
图2 T2T组装策略
错误校正:PacBio HiFi和ONT 超长读序列虽然准确,但并非完全无误。这些错误如果与遗传变异混合,可能会阻碍正确分离同源单倍型或重复序列拷贝,导致组装结果碎片化。因此,所有能够实现T2T组装的软件都会使用自己的算法来校正测序错误。
组装图构建:现代长读长序列组装软件基于图(graph-based),通过输入的读序列构建一个组装图。这个图可以是重叠图(overlap graph)或de Bruijn图(DBG)。在重叠图中,每个顶点代表一个读序列,边表示可能的连接。而de Bruijn图则是基于k-mer的,每个顶点是一个k-mer,边表示k-mer之间的重叠。这些图通常非线性,因为重复序列和倍性会导致复杂的结构。
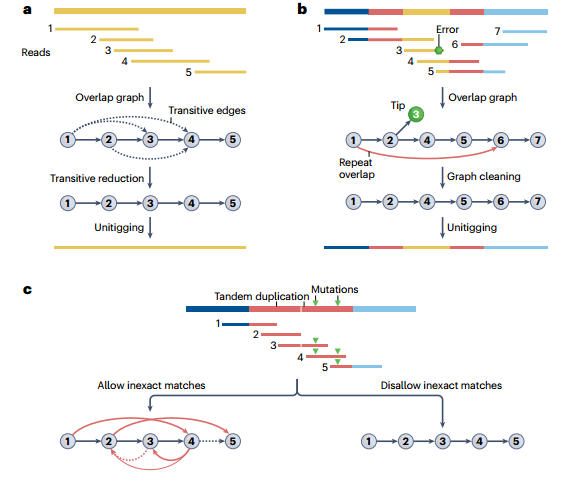
图3 基于Overlap的组装
超长读序列整合:超长读序列对于扩展分型块和组装近期复制的区域至关重要。不同的组装器使用不同的算法来整合这些读序列,以简化组装图并提高线性度。
长距离数据整合:当有来自父母的测序数据时,可以使用三重分箱算法(trio binning algorithm)实现全基因组分型。Hi-C数据对于染色体水平的分型至关重要,它通过揭示空间上接近的位点间的相互作用来辅助组装。
组装图简化:在初始组装图的基础上,需要通过各种算法进一步处理,以解决图中的复杂性并生成更长的线性序列。
支架搭建:Hi-C数据不仅用于分型,还用于在组装图的间隙处搭建支架,以提高组装的连续性。
序列polish:polishi是指通过提高序列的碱基准确性来改善组装结果的过程。对于纯合体基因组,错误的碱基不会被读序列支持,因此可以被识别和修正。
Part 3
T2T基因组的组装评估
对于一个真正的T2T基因组组装,它必须覆盖整个染色体且没有间隙,并且没有大规模的组装错误。因此,在确定组装结果是“T2T”时,必须经过严格的组装评估。T2T基因组组装评估通常包括基本指标、BUSCO评估、K-mer评估、比对评估。
基本指标:评估基因组组装的第一步通常是计算基本的统计指标。这包括组装大小(所有序列片段长度的总和)和N50(覆盖组装一半长度的最短序列片段的长度)。对于二倍体样本的常染色体,分型组装的两个基因组预期将有相似的大小。如果两个常染色体组装大小不平衡,可能表明分型不完整,可能需要手动调整参数或进行修订。
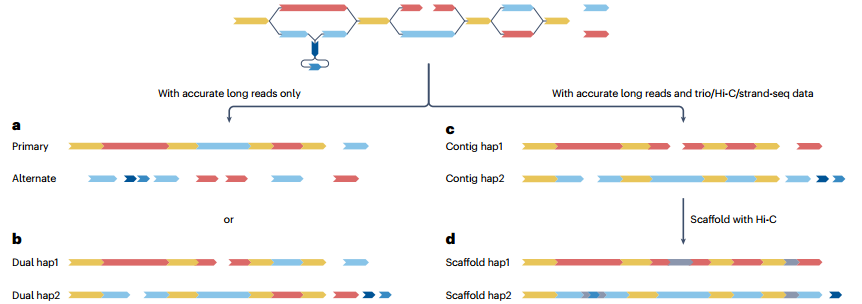
图4 基因组分型情况
基因完整性评估:BUSCO(Benchmarking Universal Single-Copy Orthologs)是一个评估组装完整性的有价值工具。它通过将保守的单拷贝蛋白与基因组对齐,并计算未对齐、断裂或重复的对齐情况来工作。未对齐的蛋白数量越多,组装的完整性越低。然而,BUSCO可能因为其对大基因组的蛋白序列与基因组准确对齐的能力有限,而低估了大型基因组的完整性。
K-mer基础评估:在均匀的读长覆盖率和完美组装的假设下,基因组组装中的k-mer计数应该与读长中的计数成比例。K-mer在组装中出现的频率高于读长表明组装中存在错误的重复,而读长中频率高但在组装中频率低的K-mer表明序列缺失。KAT是一个强大的工具,利用这些简单的观察结果来评估组装。
基于比对的评估:理想情况下,当序列读长与它们的组装对齐时,每个序列片段位置都应有均匀的覆盖率。长时间区域的覆盖率过高或过低可能表明潜在的组装错误。序列片段也应该在碱基水平上得到读长的良好支持。当从读长到组装的比对中调用变体时,孤立的小变异体可能表明序列片段共识错误,而聚集的杂合变异体可能源于折叠的串联重复。
Part 4
T2T基因组组装存在的挑战
尽管T2T组装取得了显著的进展,但目前大部分软件算法都建立在1995年建立的基本算法之上。受限于实际测序数据的特点,它们无法解决基因组中最复杂的区域,在多倍体基因组或更复杂的情况下也表现不佳,例如具有大规模拷贝数变化和重排的非整倍体癌症基因组。因此,T2T基因组组装目前还存在诸多挑战。
理论挑战:现有的两种主要组装范式——重叠图(overlap graph)和de Bruijn图(DBG)各自存在问题。在构建重叠图时,如果一个读段被更长的读段所包含,它可能会被丢弃,这可能导致组装间隙。尽管这种情况不常见,但现代组装的高度连续性使得由包含读段引起的额外组装间隙变得明显。此外,多重de Bruijn图(multiplex DBG)虽然理论上可以解决k-mer选择的困境,但实际应用中并不使用固定k-mer集合的算法,因为一套k-mer可能无法在所有子图上最优工作。
实际挑战:Hi-C数据仅提供相对分型信息,没有三重数据(trio data)的情况下,使用Hi-C作为长距离数据进行组装更为困难。当前的组装软件可能难以解决复杂的案例,如微染色体和残留的四倍体。此外,Hi-C数据的使用还有改进空间,可能通过机器学习方法进一步提升。
数据质量和实验挑战:长读长序列的生成标准协议需要大量DNA,这对于小型生物或临床样本来说可能是困难或不可能的。尽管有适用于PacBio的微量建库协议,但在低于某个阈值时,需要进行全基因组扩增,这可能引入覆盖偏差和dropouts。
多倍体和复杂基因组:尽管在二倍体基因组的T2T组装上取得了显著进展,但多倍体基因组的组装仍然是一个挑战。癌症基因组在一定程度上是多倍体的,其组装更加困难。宏基因组样本可以被视为具有更高倍性变异的多倍体基因组,其组装目标与多倍体基因组组装不同,例如,通常不期望对高度相似的基因组进行分型。
Part 5
T2T基因组组装的未来展望
由于PacBio HiFi和ONT超长reads的广泛适用性,从头组装的基因组质量在过去两年中得到了显著改善。但现在能自动组装T2T基因组中的所有染色体吗?作者认为答案通常是否定的。作者认为,过去几年的大部分进步都是由于测序数据质量的提高而取得的。目前的组装软件可以正确地重建基因组序列的长片段,但剩余的组装缺口,特别是在各种非人类基因组中,仍然难以自动修补。算法改进本身可能无法可靠地解决当前可用数据的所有基因组。期待测序技术不断取得新进展,在没有人为干预的情况下真正完成基因组。此外,完整的组装只是下游生物学发现的第一步,基因组下游的分析工具,例如全基因组比对和基因注释仍然面临重大挑战,这需要未来持续不断的进行改善。
原文链接:
https://www.nature.com/articles/s41576-024-00718-w

