
第一作者:陈 艺 研究员
通讯作者:蒲生彦 教授
通讯单位:成都理工大学
https://doi.org/10.1016/j.jhazmat.2023.132302
MnO2表面的法拉第电荷和固定电荷都有助于电极界面离子的储存;
表面法拉第过程和晶体隧道法拉第储存表现出相异的离子储存机理;
不同的法拉第和电容过程导致不同结构的MnO2具有不同的循环稳定性;
在自然CDI系统中应考虑表面法拉第赝电容行为。
MnO2作为最具潜力的法拉第电极材料,在地下水污染物去除方面具有广阔的前景。然而不同晶体结构MnO2的电容去离子动力学过程存在差异,电容-法拉第效应及多过程耦合作用机制尚不清楚。本研究通过改进Donnan 模型,结合电流-幂律关系,定量解析了目前研究广泛关注的三种晶体结构MnO2(α, β and δ)的表面和隧道中发生的电容过程与法拉第过程及其演化规律。在阴极释放电子的介导作用下,MnO2表面引发Mn4+/Mn3+法拉第反应产生大量表面法拉第电荷从而驱动重金属离子(Cd2+)的表面电荷储存(28%~47%),其次为离子嵌入赝电容过程(14%~54%)、阴极释放电子直接作用的表面电容过程(12%~34%)和晶体表面缺陷产生固定电荷储存过程(4%~8%)。Mn-O键长是决定Mn4+-O-Mn3+路径电子转移能力,产生Mn4+/Mn3+法拉第电荷的关键因素,因此表面法拉第电荷储存过程(β>α>δ)和隧道储存过程(δ>α>β)呈现一定程度的相互制约效应。本研究将原位技术与理论模拟相结合,有效阐明了MnO2电极界面的离子动力学和电荷传递机制,为拓展插层赝电容电极材料的离子储存机制的认识,形成高效稳定电容去离子电极材料调控策略提供了一个新的思路。
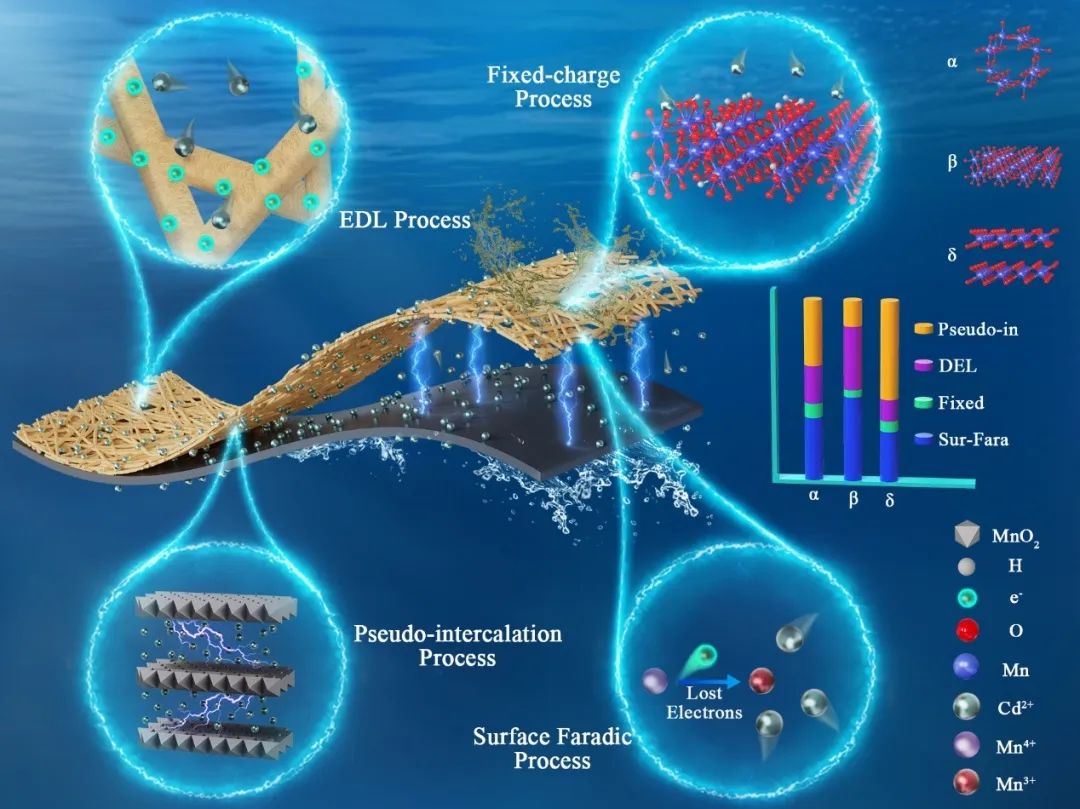
一、晶体结构驱动Cd2+的电化学吸附规律
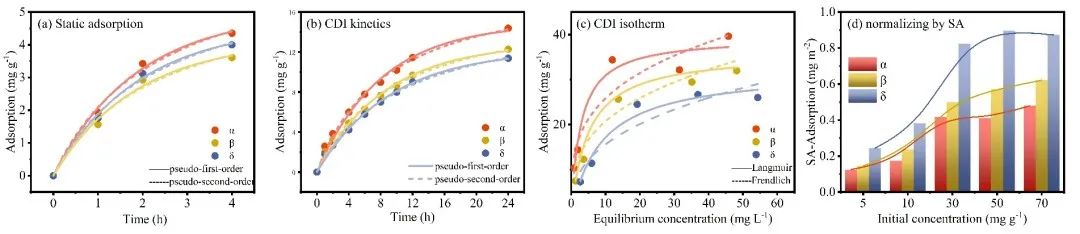
Fig 1. Electrosorption performance in different structured MnO2.
二、Cd2+离子电化学储存特征
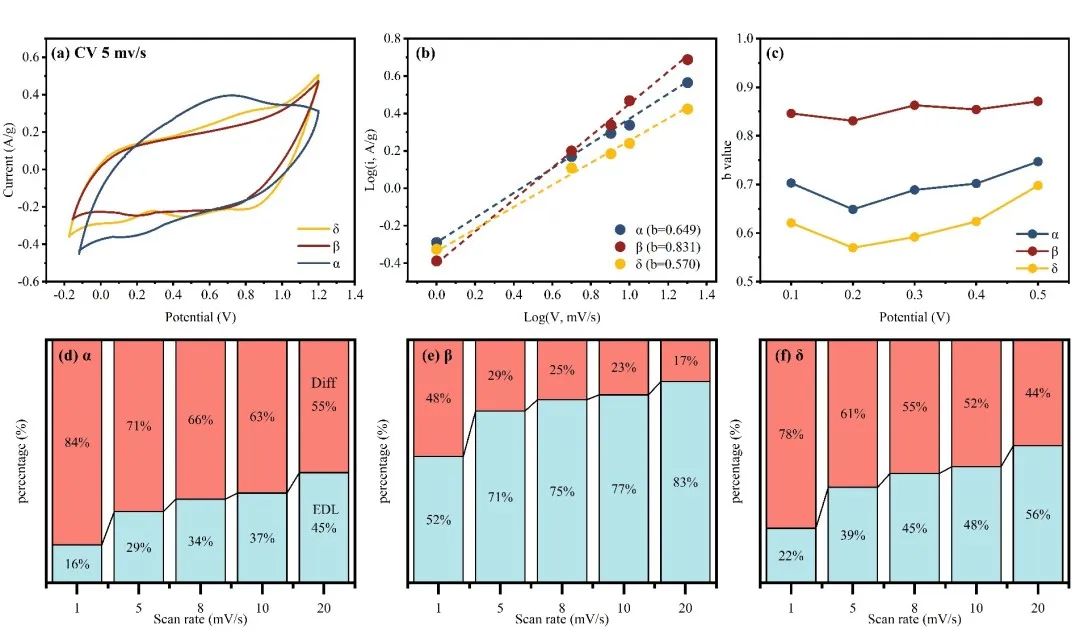
Fig 2. Electrochemical analysis. (a) CV curves of samples at 5 mV/s. (b) Plot of log (i) vs log (V) from discharge CV curves at different scan rates. (c) Calculated b values as a function of cell voltage. (d-f) Capacitance capacitive and diffusion-controlled contributions at different scan rates.
三、扩散控制法拉第嵌入过程
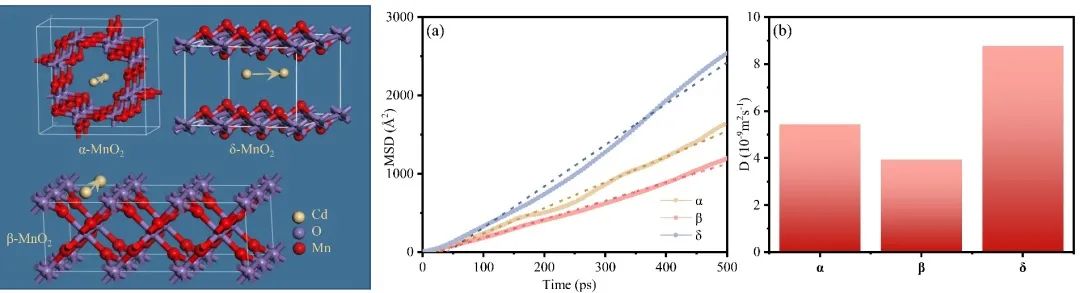
Fig 3. MSD plots and corresponding diffusion coefficients in different tunnels and layer.
四、表面固定电荷驱动离子储存过程
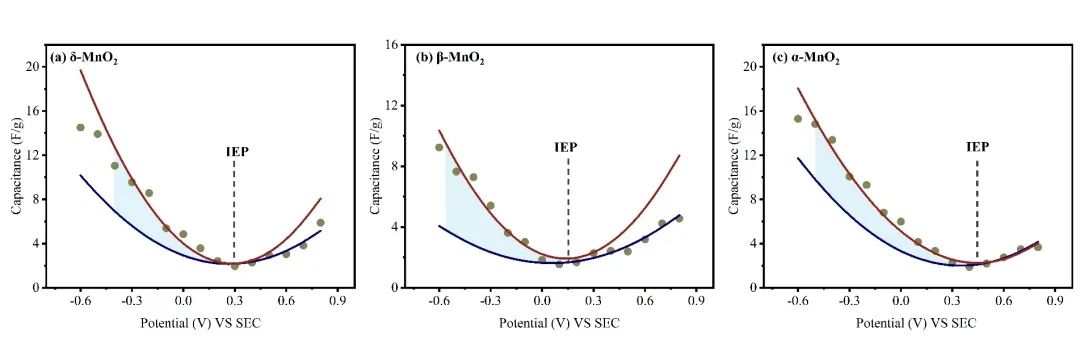
Fig 4. EIS differential capacity curves of three types MnO2 electrodes in Cd2+ solution.
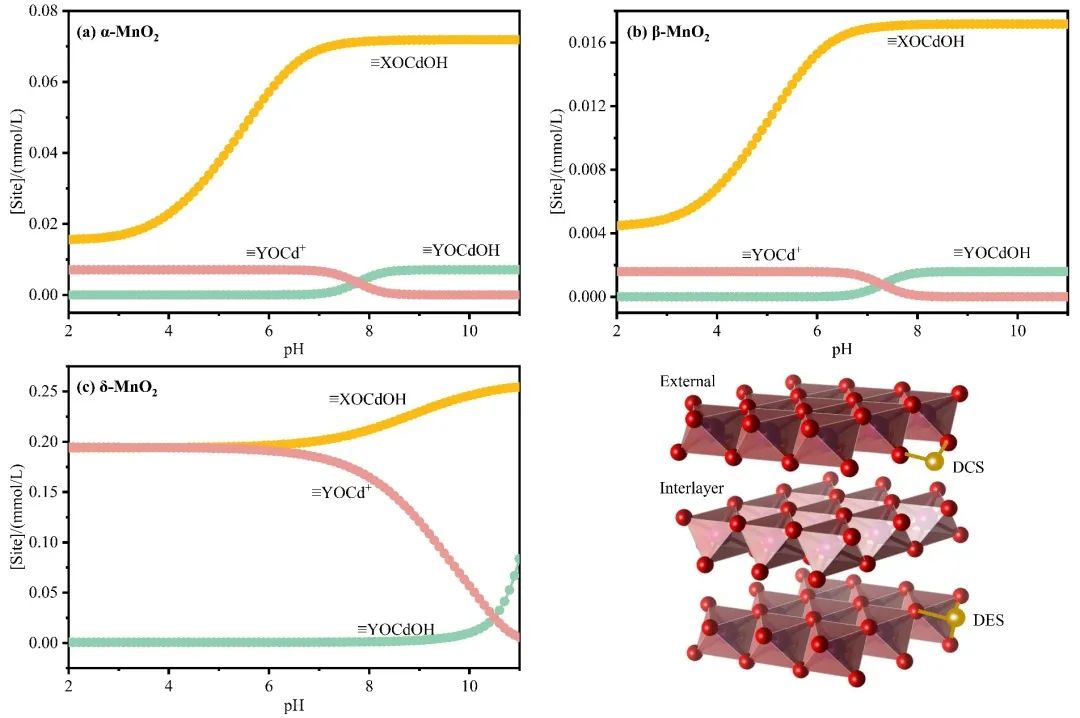
Fig 5. Modeling Cd2+ adsorption on MnO2 surface using diffuse double layer surface complexation model (SCM).
五、表面法拉第电荷驱动离子储存过程
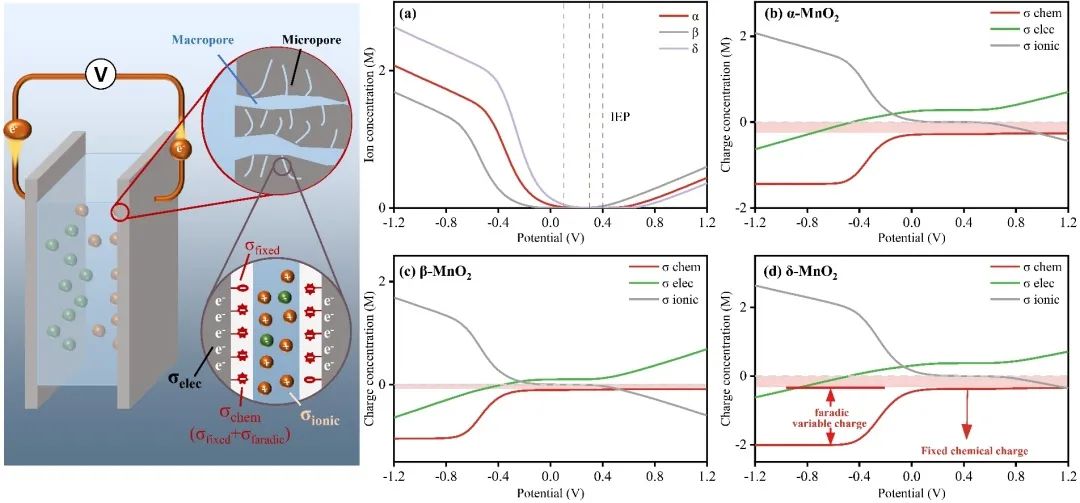
Fig 6. Modeling equilibrium concentration within micropores at various cell voltage.
本研究从离子储存过程的角度研究了MnO2的晶体结构对Cd2+电容去离子性能的影响。综合分析不同结构MnO2在不同浓度、pH和吸附情况(静态和CDI)下的吸附行为,发现晶体结构驱动效应显著(δ > β > α)。为了阐明驱动效应与晶体结构之间的结构-有效关系,我们建立了多过程耦合Donnan模型来评估特定的电荷存储行为,包括EDLC控制过程、固定电荷控制过程和法拉第电荷控制过程。与相对开放层状结构的δ相比,α和β-MnO2的隧道结构限制了离子的插层存储,但δ的晶体结构在多次吸收/解吸循环中由于在隧道中形成了O-Cd-O桥键而迅速失稳。其次,α-和δ-MnO2由于其固有的晶体缺陷或隧道异位,可以有效地增加表面吸附氧的数量和反应性,表现出明显的固定电荷控制络合行为,增强了吸附能力;阴极上相邻O2-和Mn4+之间的电子转移产生了大量法拉第电荷(Mn4+/Mn3+),促进了表面法拉第过程,因此具有较短Mn-O键的β-MnO2表现出优越的表面法拉第控制电荷吸附行为(47 %),此外,由于表面上的法拉第电荷沿Mn4+-O-Mn3+路线快速转移,表面EDL过程不像其他研究中那样占主导地位。


责任编辑:宋潇
校对丨审核:张阳 王农

欢迎各位专家学者向农业环境科学投稿宣传团队科研成果!请您联系邮箱:caep_sx@163.com。投稿时请确保您拥有论文的翻译、编辑、转载等权利。

研究揭示土壤原位微生物电场对微塑降解的强化机制电压对氧化剂和活化剂在土壤中的迁移及对PAHs去除的影响




