
伯小远在前面的推文中给大家介绍过在已知启动子的情况下如何去筛选与其结合的转录因子是什么(这里用到的是酵母单杂筛库的方法哦!),那么反过来,如果发现了一个转录因子,想要寻找与其结合的启动子是什么,该用什么方法呢?答案是ChIP-seq(染色质免疫共沉淀结合下一代测序技术),没错,小远今天将要给大家介绍的就是与染色质免疫共沉淀相关的内容,染色质免疫共沉淀实验除了可以研究转录因子与启动子的结合之外,还能做什么实验呢?想要一探究竟,就跟着小远一起往下看吧!
染色质免疫共沉淀简介
蛋白质-DNA相互作用的全基因组图谱和表观遗传标记对于充分理解转录调控是必不可少的。染色质免疫沉淀(Chromatin immunoprecipitation,ChIP)是一种在体内分离与特定蛋白结合的DNA片段的有效方法(Solomon et al., 1988)。这项技术可以将感兴趣的蛋白质与物理上和蛋白质结合的DNA片段共沉淀。DNA结合蛋白可以是转录因子(TFs)或其他染色质相关蛋白,如组蛋白。
最开始,ChIP分析通常是将分离出的DNA片段与微阵列(称为ChIP-chip)进行杂交,以确定DNA片段在全基因组中的序列。然而,随着DNA测序技术的快速发展,与DNA测序技术相关的研究日益深入 ,发展出了利用高通量测序技术对ChIP中获得的DNA片段直接进行测序的技术称为ChIP-seq。由于更高的分辨率,ChIP-seq的覆盖率和特异性相比ChIP-chip有了明显的提高(Luo and Lam, 2014)。因此,ChIP-seq已成为一种分析真核生物基因组中染色质修饰和转录因子(TF)结合位点的流行方法。
模式植物的ChIP实验流程
模式植物(水稻和拟南芥)的ChIP检测方案由Zhu等开发(Zhu et al., 2012)。首先,用甲醛处理细胞,使DNA结合蛋白在体内与DNA交联。接下来,细胞被裂解,染色质被超声波切割成小片段,然后使用特定的蛋白质抗体免疫沉淀蛋白-DNA复合物。为了保证获得高质量的数据,抗体和阴性对照的选择至关重要。市面上有许多针对表位标签的抗体,如GFP、MYC或FLAG,这些抗体通常被融合到转基因植物的蛋白中。表位标签的使用无需经过特异抗体生产的复杂过程,而野生型植株或只表达该标签的植株是完美的阴性对照。当然,ChIP也可以在野生型植株中使用针对兴趣蛋白本身的特异性抗体进行实验,这样就无需进行遗传转化实验,但是这种方法很难设计出一个完美的阴性对照,从而增加了假阳性的概率。最后一步是逆转交联,从蛋白质中释放DNA片段,然后纯化释放的DNA片段。通过实时荧光定量PCR(ChIP-qPCR)检测纯化出的DNA的质量,确定基因组感兴趣区域的富集程度。
经过上面的实验后,就到了测序和数据分析的部分。首先建一个DNA片段的测序文库,再通过测序平台进行高通量测序。测序后,将原始的ChIP-Seq数据转换为序列数据进行分析。一般来说,测序的原始读取首先通过Fastqc(www.bioinformatics.bbsrc.ac.uk/projects/fastqc/)或Fastx-toolkit(http://hannonlab.cshl.edu/fastx_toolkit/)进行质量控制,以去除接头和其它污染序列,然后使用short-read绘图程序,如Bowtie(http://bowtiebio.sourceforge.net/index.shtml),绘制到参考基因组。在成功地读取到基因组之后,下一个步骤是全基因组识别感兴趣蛋白的假定结合位点,或称为peaks calling。这个过程可以通过各种算法和程序来执行(Chaitankar et al., 2016),例如MACS (http://liulab.dfci.harvard.edu/MACS/)和QuEST (http://mendel.stanford.edu/sidowlab/downloads/quest/),以及常用于植物的CisGenome (www.biostat.jhsph.edu/~hji/cisgenome/)。最后,结合序列基序的分析可以通过MEME、 DREME或MEME-chip来完成(http://memesuite.org/)。
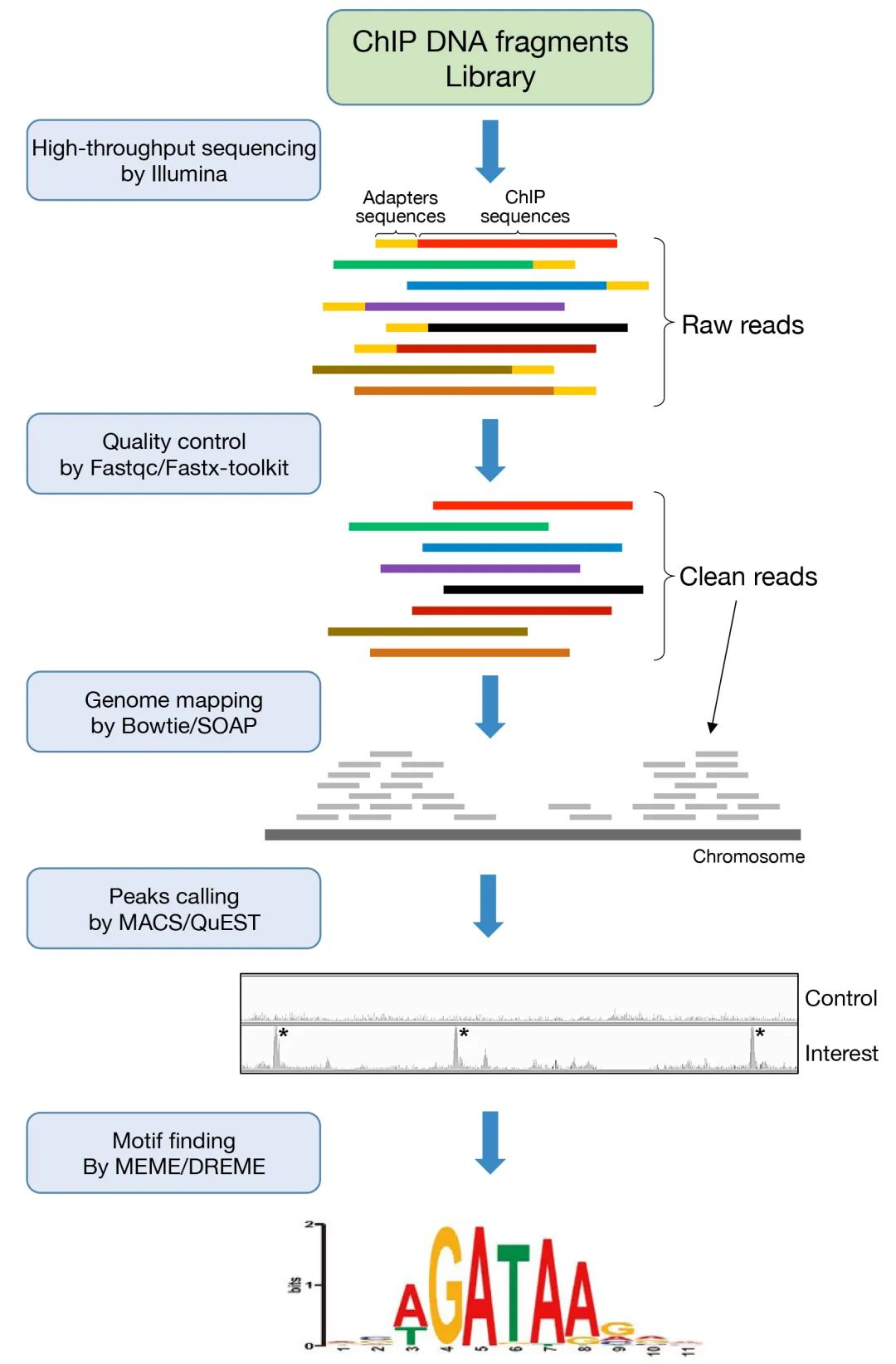
图1 ChIP-seq数据分析工作流程概述(Chen et al., 2018)。
这里给大家介绍几个专业名词:
Peak calling:查找DNA结合位点的步骤一般叫做peak calling。TF在基因组上的结合其实是一个随机过程,基因组的每个位置其实都有机会结合某个TF,只是概率不一样,说白了,peaks出现的位置,是TF结合的热点,而peak calling就是为了找到这些热点。如何定义热点呢?通俗地讲,热点是这样一些位置,这些位置多次被测得的reads所覆盖(测序时一般测的都是一个细胞群体,reads出现次数多,说明该位置被TF结合的几率大)。换句简单的话说,你可以理解peaks就是reads峰。
Peak注释:所谓的peaks注释,首先看peaks在基因组的哪一个区段,看看它们在基因不同区域(基因上下游,5’/3’-UTR,启动子,内含子)的分布情况。最典型的对于转录因子,通常都是位于基因的启动子区;其次是邻近的基因的注释,蛋白结合到DNA上之后,主要是发挥基因表达调控的功能,这些peaks区域附近的基因就作为其候选的调控基因。
Motif(基序)分析:在ChIP-seq数据分析中,motif分析是一项重要的分析内容。通过motif分析,我们可以对转录因子结合位点的序列模式有进一步的了解,那么什么是motif呢?蛋白质发挥功能的基本单元是domain,是一种特殊的三维结构,不同结构的domain与其他分子特异结合从而发挥功能。与此类似,转录因子在与DNA序列结合时,其结合位点的序列也由于一定的特异性,不同转录因子结合的DNA序列的模式是不同的。为了更好的描述结合位点序列的模式,科学家们提出了motif的概念——特定碱基序列的模式即为motif。
下面是我们在文章中或motif分析中经常见到的图,用来表征序列的一致性和多样性。字母越大,代表在该位置出现该核苷酸或者氨基酸的概率越大,常用Bits或者百分比表示。

图2 motif示例(Chen et al., 2018)。
注:一般情况下一个转录因子在多个基因上的结合序列是不同的,用motif表示这个信息时涉及到一些其它的概念,例如碱基分布频率、一致性序列等,这里小远就不给大家一一展开了,有兴趣的同学可以自己去查看资料哦!
为了加深大家对ChIP-seq的理解,下面再从实验本身的角度为大家讲解一下这个实验的过程,希望通过伯小远的反复讲解大家对这个实验可以有一个比较透彻的理解,等理解完这个实验,我们再去看它的应用!
ChIP-seq一般实验过程
样本预处理:收获植株后,植物组织立即固定,以交联蛋白质-DNA相互作用。甲醛是最常用的交联剂,交联完成后加入甘氨酸以终止反应。注意:交联剂在整个过程中保持复合物稳定,但其作用必须是可逆的,这样才能用于ChIP。
获取核DNA:裂解细胞,得到全细胞的裂解液,提取核DNA。
DNA片段化:DNA片段化是获得良好的ChIP分辨率的关键因素,理想情况下的片段大小在200到1000bp之间。剪切是最难控制的步骤之一。通过超声处理和/或核酸酶/酶促消化可以实现剪切,各有利弊。超声处理虽然需要大量的手动操作,但非常适合难以裂解的细胞;酶促消化不需要手动操作,适用于大量样品的处理,但其剪切位点不是随机的。
免疫沉淀:抗体免疫沉淀,实验组加入预先结合了特异性抗体的beads进行孵育,最终形成beads-抗体-目的蛋白-DNA复合物。
解交联:洗脱得到抗体-目的蛋白-DNA复合物,去除非特异性结合的DNA片段,使用蛋白酶K/NaCl处理进行解交联,最后将DNA片段纯化回收。
验证:通过qPCR对ChIP结果进行验证。
高通量测序:准备好ChIP后的DNA样品用于ChIP-seq建库,质检及测序。

图3 染色质免疫沉淀ChIP-seq/qPCR实验流程。
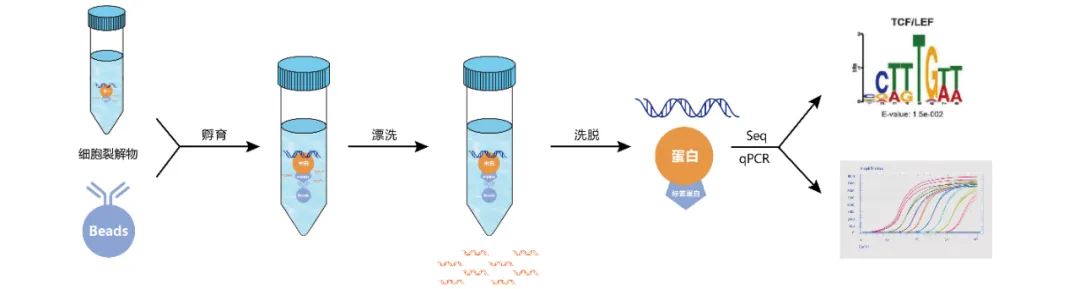
图4 带标签的染色质免疫共沉淀流程图。
ChIP-seq的应用
上面对ChIP-seq的一些基本概念以及实验流程进行了介绍,那么介绍完这个实验之后,这个实验除了本文开头提到的寻找与转录因子结合的启动子之外,还可以用来干什么呢?以下就是ChIP-seq的几个主要应用方向:
1、ChIP-seq可以研究组蛋白的修饰情况,以剖析表观遗传特征和生物学功能;
2、 ChIP-seq可用来研究转录因子结合位点,解析该转录因子作用的通路信息;
3、ChIP-seq技术可得到核小体的定位图谱,核小体定位在转录调控,DNA复制和修复等多种细胞过程中并起着重要作用;
4、ChIP-seq技术可研究DNA的甲基化情况,DNA甲基化会引起染色体结构、DNA构象、DNA稳定性以及DNA与蛋白质相互作用方式的改变,从而控制基因表达。
针对ChIP-seq这4个方面的应用,为了让大家能更好地理解文献举例里面的内容,小远决定在这里先给大家把基础打牢,如果不清楚基本概念,那么有些实验结果可能会比较难理解,所以放慢脚步看看下面都有些什么内容需要我们提前学习吧!
表观遗传学
固着陆地植物已经进化出了对各种环境信号作出反应的能力。许多研究报告已经确定了植物中调控胁迫反应的分子成分(转录因子、转运体、信号转导等)。后来,一种可逆的表观遗传调控系统被报道具有协调不同生命过程的基因组、转录、翻译和代谢反应的能力(Kouzarides, 2007;Choudhary et al., 2014;Holoch and Moazed, 2015;Kinnaird et al., 2016)。一般来说,表观遗传调控的作用模式在真核生物中是高度保守的。因此,表观遗传调控是各种生物过程的基本组成部分(Allis and Jenuwein, 2016)。对非生物胁迫响应的表观遗传调控的研究,揭示了由表观遗传元素协调的植物胁迫响应的部分或完整的图景(Kim et al., 2015;Asensi-Fabado et al., 2017;Luo et al., 2017)。组蛋白修饰、组蛋白变异、染色质重塑、调节RNA(如非编码RNA)和DNA甲基化都是表观遗传调控的元素(Goldbergetal.,2007)。
简而言之,表观遗传学其实就是研究在没有细胞核DNA序列改变的情况时,基因功能的可逆的可遗传的改变。也就是说,在不改变基因组序列的前提下,通过DNA和组蛋白的修饰等来调控基因的表达。
染色质的结构
染色质的基本重复结构(和功能)单位是核小体,核小体包含8个组蛋白和大约146个碱基对的DNA (Van Holde, 1988;Wolffe, 1999)。电子显微镜下观察到的染色质类似于串珠,为核小体的存在提供了早期线索(Olins and Olins, 1974;Woodcock et al., 1976)。另一个线索来自于染色质中的组蛋白的化学交(即连接)(Thomas & Kornberg, 1975)。实验证明H2A、H2B、H3、H4形成了一个离散的蛋白质八聚体,这与染色质纤维中存在重复的组蛋白单元完全一致。
组蛋白是一个小的带正电的蛋白质家族,被称为H1, H2A, H2B, H3和H4 (Van Holde, 1988)。DNA是带负电荷的,因为它的磷酸糖主干上有磷酸基团,所以组蛋白与DNA结合非常紧密。
核小体的结构如下:两个组蛋白H2A、H2B、H3和H4聚在一起形成一个组蛋白八聚体,它结合和包裹大约1.7圈的DNA,或约146个碱基对。一个H1蛋白的加入包裹了另外20个碱基对,导致八聚体发生了两次完整的旋转,形成了一个称为染色体的结构(图6中的Box 4)。考虑到平均每条染色体包含1亿多对DNA碱基对,由此产生的166个碱基对并不算长。因此,每条染色体包含数十万个核小体,这些核小体由它们之间的DNA连接(平均约20个碱基对)。这个连接的DNA被称为连接DNA。因此,每条染色体都是一个长链的核小体,当用电子显微镜观察时,它看起来像一串珠子(图4)(Olins & Olins, 1974, 2003)。

图5 染色质的电子显微图:串珠(Olins D E and Olins A L, 2003)。

图6 核小体结构。(图片来源:https://www.jieandze1314.com/post/cnposts/191/)
每个核小体的DNA数量是通过用一种切割DNA的酶(这种酶被称为DNA酶)来处理染色质来确定的。其中一种酶,微球菌核酸酶(MNase),具有重要的特性,即在切割包裹在八聚体周围的DNA之前,优先切割核小体之间的连接DNA。通过调节应用MNase后发生的切割量,有可能在每个连接DNA被裂解之前停止反应。此时,处理过的染色质将由单核小体、双核小体(通过连接物DNA连接)、三核小体等组成 (Hewish and Burgoyne, 1973)。如果MNase处理的染色质的DNA在凝胶上分离,就会出现许多条带,每个条带的长度是单核小体DNA的倍数(Noll, 1974)。对这一观察结果最简单的解释是,染色质具有基本的重复结构。晶体学家根据他们的数据构建的核小体模型如图7所示。DNA双螺旋的磷酸二酯骨架显示为棕色和绿松石色,而组蛋白显示为蓝色(H3)、绿色(H4)、黄色(H2A)和红色(H2B)。注意,只有真核生物(即具有细胞核和核膜的生物)才有核小体。原核生物,如细菌,则没有。
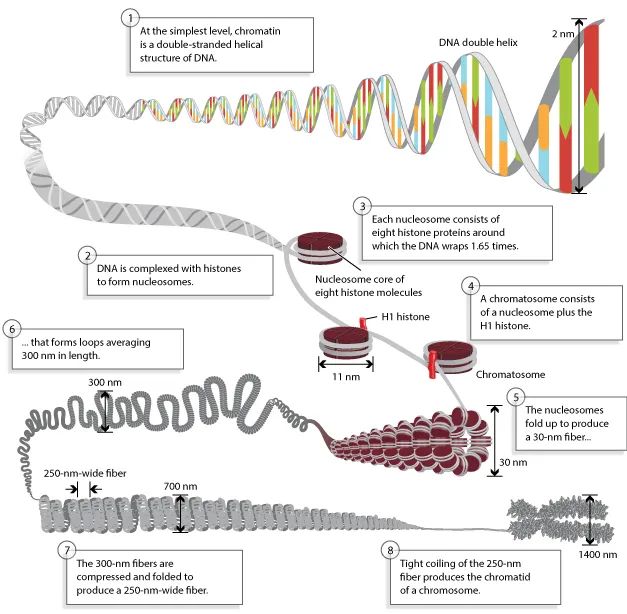
图7 染色体是由紧密缠绕在组蛋白上的DNA组成的(Annunziato A, 2008)。在组蛋白的帮助下,染色体DNA被包裹在微小的细胞核内。这些是带正电荷的蛋白质,它们强烈地粘附在带负电荷的DNA上,形成被称为核小体的复合物。每个核小体由8个组蛋白缠绕1.65圈的DNA组成。核小体折叠形成一个30nm的染色质纤维,形成平均300nm长的环。300nm的纤维被压缩和折叠,产生一个250nm宽的纤维,该纤维紧紧地盘绕在染色体的染色单体中。
图中的8个过程:
①在最简单的水平上,染色质是一个双链螺旋结构的DNA。
②DNA与组蛋白结合形成核小体。
③每个核小体由8个组蛋白组成,DNA围绕组蛋白包裹1.65圈。
④染色体由核小体加上H1组蛋白组成。
⑤核小体折叠形成30nm的染色质丝。
⑥30nm染色质丝平均每300nm盘曲成环。
⑦300nm的纤维被压缩和折叠,产生一个250nm宽的纤维。
⑧250nm纤维的紧密结合产生了一个染色体的染色单体。

图8 核小体核心颗粒:146bp DNA磷酸二酯骨架(棕色和绿松石色)和8个组蛋白主链(蓝色:H3,绿色:H4,黄色:H2A,红色:H2B)。
组蛋白修饰
今天给大家介绍的都是比较基础的内容,重在为大家打基础,虽然开头是以ChIP-seq可以寻找与目的转录因子结合的启动子是什么开始的,但讲到最后也没见到相关的实例,这个大家可以先别着急,后面的文章中会为大家讲到,耐心期待一下哦!ChIP-seq除了研究转录因子与启动子的结合,还有很多其它的应用,特别是表观遗传学,所以对于这一块的内容小远就稍微多讲了点,希望对大家有所帮助,不要嫌伯小远啰嗦。好了,本期的文章到这里结束了,后面再为大家介绍ChIP-seq的应用实例!


