项目概述
2021年发表在BioRxiv期刊上题为“The complete sequence of a human genome”的文章利用PacBio HiFi和Nanopore测序技术,构建了首个真正意义上的无gap智人基因组图谱,同时利用多种数据类型对基因组进行了评估。
文章的主要工作如下:
01. 总结了人基因组组装的主要版本:2013的GRC和2019年gap filling的GRCh38.p13;
02. 简述了本次组装的无gap的T2T-CHM13的组装策略及组装过程;
03. 比较了本次无gap的基因组组装版本较之前版本的提升,主要体现在:
校正了大量的碱基错误,发现了约200M的新序列,包含2226个paralogous gene copies,以及115个编码蛋白;
组装提升的主要区域包括着丝粒区域以及5个近端粒的短臂;
首次解析了这些复杂区域的结构及功能研究。
测序平台及数据
文章中采用的数据类型及测序深度统计如下:

组装策略
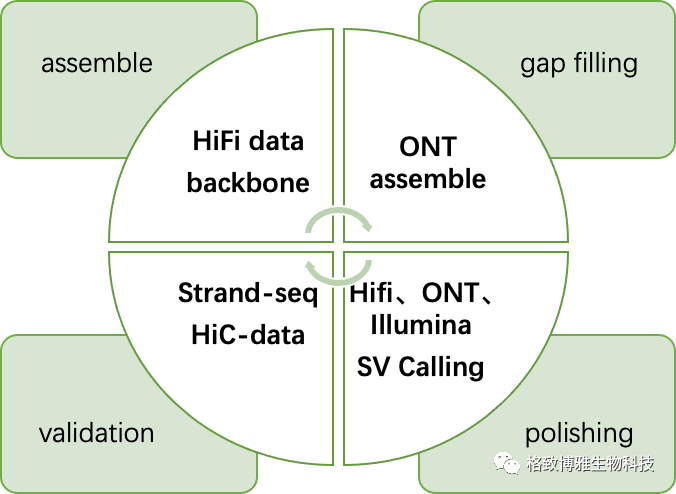
用HiFi reads构建基因组骨架,ONT数据用于gap filling,目标区域主要针对高重复的端粒区域。
用其他不同类型的数据进行回比,SV calling,对基因组骨架进行校正,同时验证SV的准确性,进行基因组的评估。
通过重复以上流程,不断进行各类型数据的比对及基因组组装结果的校正过程,最终将rDNA arrays序列合并到校正后的基因组中,获得telomere-to-telomere representation of a human genome,过程如下图所示:
01
基因组组装
利用HiCanu和Miniasm对HiFi reads进行初步组装,构建基因组的骨架。具体步骤如下:
①同聚物压缩,有研究表明同聚物长度估算错误是最主要的HiFi错误模式。采用同聚物压缩,将相同的核苷酸压缩为1个碱基,比如'AAA...'->'A';
②对压缩的同聚物进行比对和纠错,同时重新计算纠错后的overlap;
③根据比对的结果进行mask,提取具有1kb的overlap做组装图形的构建,利用Miniasm的(Myers string graph)算法进行基因组的构建;
02
补洞及纠错
补洞
利用winnowmap v1.1软件,将ONT Ultra-long reads比对到已组装的基因组,对初步组装的基因组进行gap的从头填充,补丁的位置通过minimap2的比对来确认。
命令行为:
minimap2 -H -x asm20纠错
SV calling,对于长reads及短reads分别采用了不同的sv calling策略。
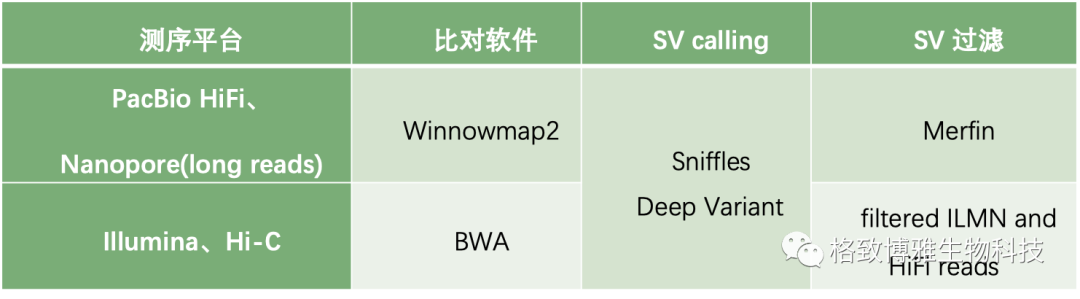
主要命令行如下:
sniffles -s 3 -d 500 -n -1 bamJasmine v1.0.2 max_dist=500 min_seq_id=0.3 spec_reads=3 --output_genotypes将过滤及验证后的SV定位到初步组装的基因组骨架下,对该部分进行纠错。
03
组装结果验证
利用不同平台的数据对纠错后的基因组进行验证。
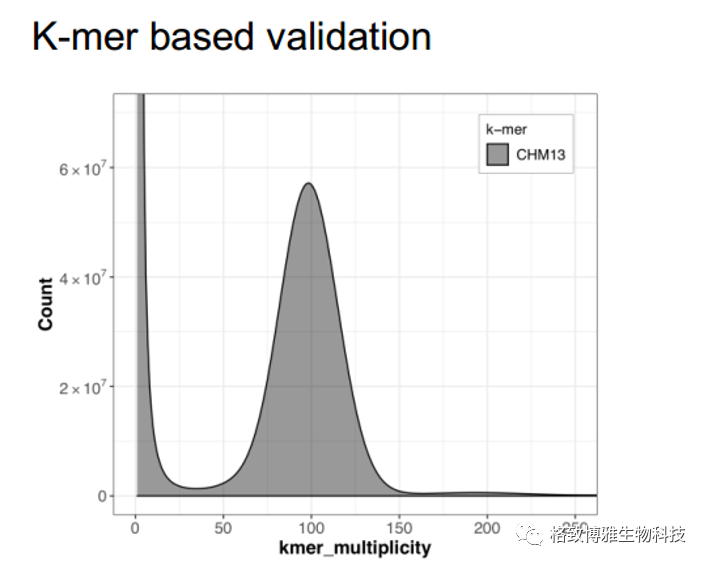
Illumina数据评估
利用软件Merqury(githup: https://github.com/marbl/merqury),通过切kmer比对基因组的方式,获得二代数据对基因组一致性的质量值,质量值越高,表明组装基因组的准确性越高。
HiFi和Nanopore数据评估
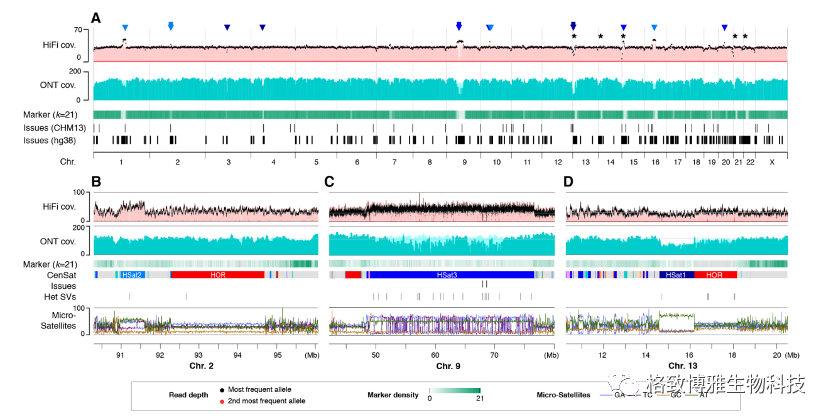
利用Winnowmap2将HiFi和Nanopore数据比对到纠错后的基因组,利用IGV查看产生填充GAP和SV的区域的覆盖深度,对纠错后的基因组进行评估。
Hi-C数据评估
将纠错后的基因组进行HIC辅助组装,主要评估端粒区域及着丝粒区域,软件可视化采用PretextView。
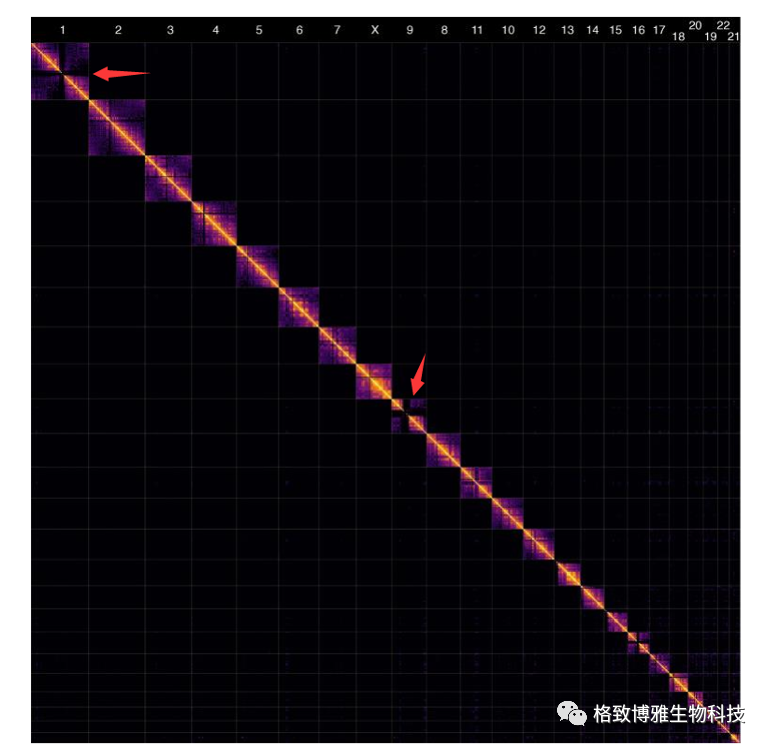
基因组组装结果一致性评估
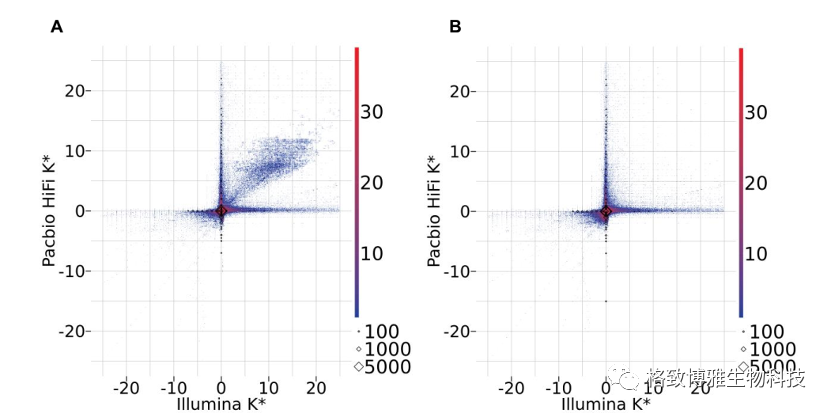
利用软件: Merfin k* statistics的统计模式,通过三代数据和二代数据的一致性的比对方式,对基因组进行了过滤处理,获得过滤后的基因组序列。当Illumina数据和HiFi数据对基因组的评估达到完全一致时,获得最终的基因组组装结果。
原文链接:https://doi.org/10.1101/2021.05.26.445798

